随着全球经济及社会的发展,人们对于能源的需求及使用日益增长。环境污染和化石能源匮乏的问题日益显著,为了人类的可持续发展。寻求开发新能源和可再生资源迫在眉睫。太阳能和风能等新型能源虽然便利清洁,但是由于其自身受时空分布不均匀的特点限制在现阶段并不能广泛使用。
作为化学储能装置,锂离子电池以比功率高、能量密度大、寿命长、自放电率低和贮藏时间长等优点,被广泛应用于便携式电子设备、航天、军事装备及电动交通工具。目前,锂离子电池已逐步替代其他电池为主要的动力电池。另一方面,由于近年来智能电网及大规模储能领域的发展对锂离子电池的能量密度和功率密度提出了更高的要求,这使得开发具有高能量密度和大功率密度的新型锂离子电池尤为重要。
第一性原理计算方法即从头算(ab initio)被广泛应用在化学、物理、生命科学和材料学等领域。它的基本思想是将多个原子构成的体系看成是由多个电子和原子核组成的系统,并根据量子力学的基本原理对问题进行最大限度的“非经验性”处理。它只需要5个基本常数(m0,e,h,c,kB)就可以计算出体系的能量和电子结构等物理性质。
第一性原理计算可以确定已知材料的结构和基础性质,并实现原子级别的精准控制,是现阶段解决实验理论问题和预测新材料结构性能的有力工具。并且,第一性原理计算不需要开展真实的实验,极大地节省了实验成本,现已被广泛应用于锂离子电池电极材料的嵌脱锂机理探索、扩散能垒计算、结构稳定性、嵌锂容量机理研究等方面,为锂离子电池电极材料的制备和改性提供了有效的理论指导。
其中,在锂电领域,利用第一性原理计算为锂离子电池材料的设计提供的理论应用主要集中于以下几个方面:
1 工作电压的计算
锂离子嵌入电压是锂离子电池的一个重要参数,而理想的材料是正极材料的电压平台足够高、负极材料的电压平台足够低,才能得到较高的工作电压,进而为锂离子电池提供较高的能量密度。第一性原理可以通过计算材料基态的电子总能量计算出平均嵌锂电压(average intercalationvoltage,AIV),与实验测到的电压数值比较接近,其原理阐述如下,例如电极反应式:
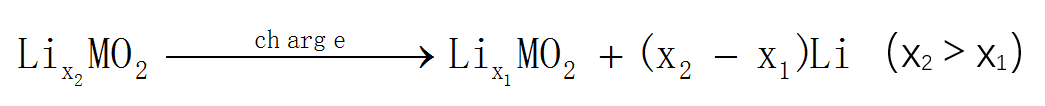
其开路电压可由如下公式计算所得:

其中,μcathode和μanode分别为锂原子在正负极材料中的化学势,z为反应过程中转移电子数,F是法拉第常数,△G为吉布斯(Gibbs)自由能。
在0K时,可近似为△G≈△E,则公式1可写为:
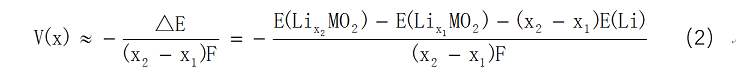
因此,只要计算反应前后的各物质的总能量,就可以利用公式(2)求解正极材料的平均电压。第一性原理计算可以比较准确地预测材料的平均嵌锂电压,与实验测到的电压数值比较接近,如Zhou等2人通过计算得正极材料LiNiPO4的电压为5.1V,而实验测试值为5.1V-5.3V。Chen等3通过计算所得正极材料LiFePO4的平均电压为3.2V,其实验值为约3.4V。另外, Hassan等4利用第一性原理计算所得到的RuO2负极材料工作电压曲线,与实验中所获得工作电压曲线变化趋势定性的符合。
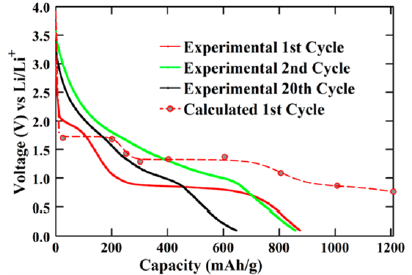
2 电子传导性和离子扩散性
倍率性能是指电池在一定时间内放出其额定电容的电流值。倍率性能越高的电池,放出相同容量的时间则越短,这有利于电池快速的充放电。材料的离子电导率和电子电导率共同影响着材料的倍率性能。高倍率下的充放过程不仅需要快速的离子扩散,也需要快速的电子传导。
利用第一性原理计算的方法,可以采用NEB(Nudged elastic band)和CI-NEB(A Climbing image nudged elastic band)的方法,对材料中锂离子的扩散能垒进行计算,而扩散能垒则对应着锂离子的扩散能,也就是扩散速率。扩散能垒越低的材料,其扩散速率越大,则相应的倍率性能则越高。像大家在文献中所看到的诸如此类的扩散能垒图5,都是通过第一性原理计算的方法进行计算的。N掺杂石墨烯能够改善负极材料的锂离子扩散速率,除了在实验中测得的实验值来验证外,也可以通过第一性原理计算来计算不加N掺杂石墨烯时材料中锂离子的扩散能垒,通过和加N掺杂石墨烯后的复合材料的锂离子扩散能垒进行对比来分析复合材料中扩散能垒的降低是否真的是引入N掺杂石墨烯引起的。
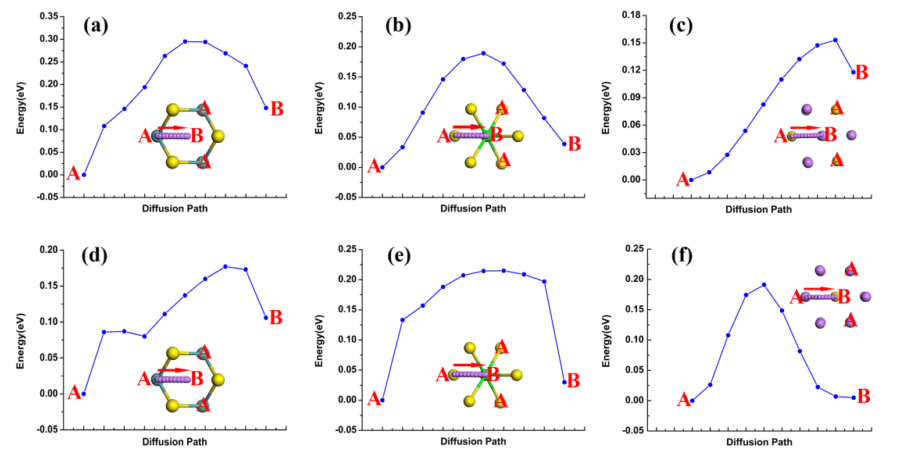
3 材料结构稳定性的计算
安全性能一直是锂离子电池的一个重要指标,这影响了电极材料和电解液的选择,我国曾出现过车载锂离子电池起火的事故,正是因为电池在使用过程中造成短路导致的。所以,必须选择结构和热稳定性均良好的材料作为锂离子电池的电极材料。在锂离子电池正极材料充放电循环中,在深度脱锂时,正极材料可能会释放O2,这不仅会消耗电解液,更会导致爆炸,造成重大安全问题。
利用第一性原理计算,可以通过计算材料缺陷的形成能和迁移能,来预测相稳定性。例如Hakim Iddir等6基于第一性原理计算,通过计算Co空位的形成能和迁移能,预测了xLi2MnO3?(1?x)LiMO2的相稳定性。Gao等7基于DFT和FPMD分析了Ti,V,Cr,Fe,Co,Ni,Zr和Nb等元素掺杂Li2MnO3材料中的Mn对于材料性能的影响,通过定义O的反应焓,计算吉布斯自由能,来研究掺杂后材料中O2生成的难易程度。Ti-,V-,Cr-,Co-,Ni-和Zr-doped在含Li量y=1.5之前达到零点,因此,其掺杂不能推迟O2的释放。而Fe-和Nb-doped在Li移除量超过0.5时仍未达到零点,表明其掺杂可以抑制材料在反应中的O2的生成,从而使得材料的安全性能得到提升,其理论计算的结果与实验掺杂得到的结果一致。
4 储锂容量的计算
电极材料的容量是电极中非常重要的性能,在第一性原理计算中,可以通过电极材料对锂原子的吸附能来进行容量的分析。吸附能的大小可以比较不同材料对锂原子的吸附能力,吸附能越大的材料,其吸附锂原子的能力则越强。但是,吸附能大的材料,其容量并不一定高。因为吸附能越大,如果其继续吸附锂原子后,吸附能降低的速率很大的话,那么这种材料的储锂容量便不会高。如果吸附能越大,当逐渐增加锂原子后吸附能的降低速率也很平缓时,这种材料就有可能拥有较大的储锂容量。锂原子有内聚能,也就是锂原子自身形成锂块体时所对应的能量。当锂原子在材料中的吸附能低于内聚能时,这时锂原子倾向于形成锂块体,而不再为电池的容量做贡献,也就是说,当我们利用第一性原理计算得到材料的吸附能低于锂块体的内聚能时,此时所对应的的储锂容量则为该材料的理论储锂容量。
例如Wang等8利用第一性原理计算得到了(掺杂)石墨烯与金属氧化物负极材料的反应产物Li2O构成的界面储锂容量,为金属氧化物在实验中所观察到的额外容量的产生提供了机理的解释。
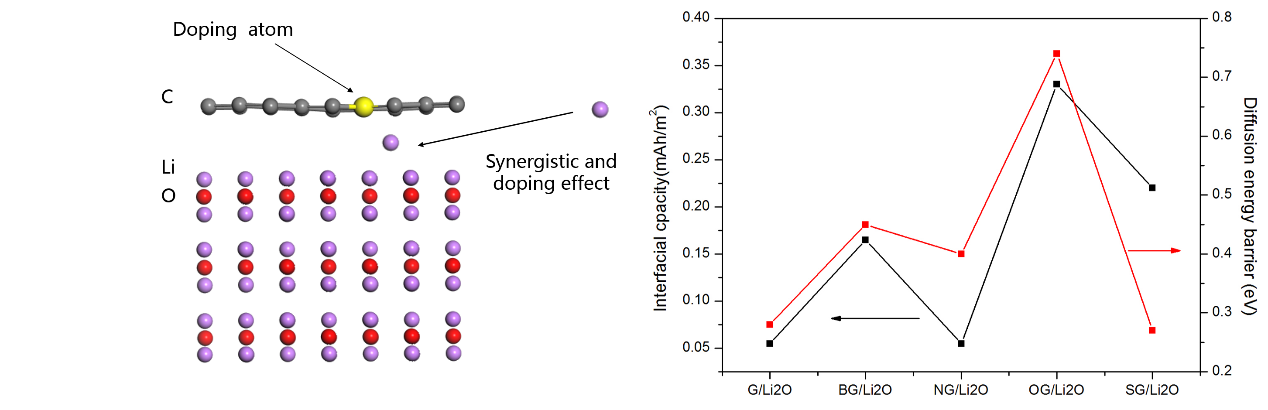
但是,第一性原理计算在现阶段锂离子电池领域中的应用也有局限性,因为实际电极材料的工作状态是在多种反应共存的条件下进行的,而通过第一性原理计算模拟的材料性能是在理想的平衡态条件进行的,这可能造成计算值与实验值产生一定的偏差。但是,通过第一性原理计算得到的数值可以定性的帮助实验工作者进行辅助分析,解释实验中存在的一些机理问题,为锂离子电池电极材料的设计提供一定的帮助。
最后,给大家进行一个简单的词汇科普—VASP。大家看到的在锂电领域第一性原理计算的文献中经常所看到的VASP一词,其实是Vienna Ab-intio Simulation Package的缩写,它是基于密度泛函理论并利用平面波赝势方法进行从头分子动力学和第一性原理计算电子结构计算的软件包,是目前材料模拟和计算材料科学研究中非常流行的商用软件。Vasp软件是由J. Furthmuller和G. Kresse首先开发和利用的,并在后期得到了不断的更新和完善,如今使用的Vasp软件包已相当成熟。
Vasp软件包具有以下优点:
(1)它给出了周期表中几乎全部元素的赝势,这些赝势已经经过充分的测试,形成了一个可用性非常高的赝势库。
(2)优化算法的实现(RMM-DISS,blocked Davidson和共轭梯度算法)效率高、稳定性好。
(3)虽然没有图形界面,但是使用文档详细,入门快。
(4)所支持的计算机平台(单机,计算集群,超级标量计算机和超级向量计算机)非常广泛,几乎在所有架构(Intel的Pentium系列、Athlon系列的CPU、DEC的Alpha机等等)的计算机器的运行效率都非常高。